?unique=a022b2a)
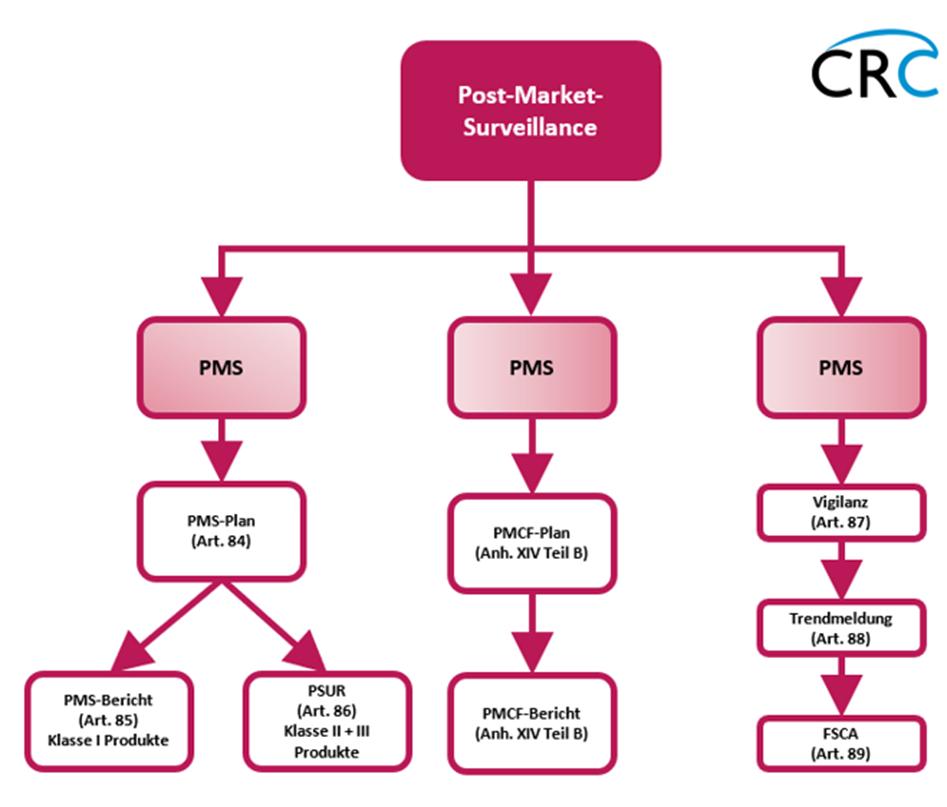
Die unten dargestellte Abbildung verschafft eine Übersicht zu den Anforderungen, die die MDR an den PMS-Prozess stellt. Ein Medizinprodukte-Hersteller muss in einer Weise, die hinsichtlich der Risikoklasse und Art des Produktes angemessen ist, ein System zur Überwachung nach dem Inverkehrbringen planen, einrichten, dokumentieren, anwenden, Instand halten und auf den neuesten Stand bringen. Der Hersteller ist dabei verpflichtet, einschlägige Daten über die Qualität, Leistung und Sicherheit eines Medizinprodukts während der gesamten Lebensdauer zu sammeln, aufzuzeichnen und zu analysieren, sowie erforderliche Schlussfolgerungen zu ziehen und etwaige Präventiv- oder Korrekturmaßnahmen zu ermitteln und zu überwachen.
Die Überwachung nach dem Inverkehrbringen fordert einen PMS-Plan, in dem der Hersteller den Nachweis über die Erfüllung seiner Verpflichtungen gemäß Artikel 83 erbringt. In einem PMS-Bericht fasst der Medizinprodukte-Hersteller die Ergebnisse und Schlussfolgerungen der Analysen mit einer Begründung und Beschreibung etwaiger Präventiv- und Korrekturmaßnahmen zusammen. Der PMS-Bericht gilt für Medizinprodukte der Klasse I. Für Medizinprodukte der Klassen 2a, 2b und 3 wird der PSUR-Bericht (Periodic Safety Update Report) erstellt. Im Vergleich zum PMS-Bericht umfasst der PSUR-Bericht umfangreichere Daten., wie zum Beispiel Schlussfolgerungen aus der Nutzen-Risiko-Abwägung oder Informationen zur gesamten Absatzmenge des Produkts.
Der PMS-Prozess umfasst zudem eine klinische Nachbeobachtung nach dem Inverkehrbringen (PMCF Post Market Clinical Follow-Up), welche einen fortlaufenden Prozess zur Aktualisierung der klinischen Bewertung gem. Artikel 61 und Anhang XIV Teil A der MDR darstellt. Außerdem wird das PMCF im PMS-Plan behandelt. Das PMCF basiert auf einem PMCF-Plan und einem PMCF-Bericht. Sowohl der PMCF-Plan als auch der PMCF-Bericht werden in Anhang XIV Teil B der MDR behandelt.
Ebenso sind Vigilanz, die Meldung von schwerwiegenden Vorkommnissen, Sicherheitskorrekturmaßnahmen im Feld (FSCA-Meldungen) und die Meldung von Trends Teil des PMS. Das Vigilanzsystem ist ein Meldesystem, über das Medizinprodukte-Hersteller schwerwiegende Vorkommnisse zu ihren Medizinprodukten den zuständigen Behörden über die Europäische Datenbank für Medizinprodukte (EUDAMED, gem. Artikel 92) melden und gegebenenfalls FSCAs ergreifen.
Daten aus dem PMS-Prozess werden für folgende Zwecke verwendet:
-
- Aktualisierung der Nutzen-Risiko-Abwägung und Verbesserung des Risikomanagements gem. Anhang I Kapitel I,
-
- Aktualisierung der Auslegung und der Informationen zur Herstellung, Gebrauchsanweisung und Kennzeichnung,
-
- Aktualisierung der klinischen Bewertung,
-
- Aktualisierung des Kurzberichts über Sicherheit und klinische Leistungen gem. Artikel 32,
-
- Ermittlung des Bedarfs an Präventiv-, Korrektur- oder Sicherheitskorrekturmaßnahmen,
-
- Ermittlung von Möglichkeiten zur Verbesserung der Gebrauchstauglichkeit, der Leistung und Sicherheit ggf. als Beitrag zur Überwachung anderer Produkte nach dem Inverkehrbringen und
-
- Erkennung und Meldung von Trends gem. Artikel 88.
Nur durch die Sicherstellung einer kontinuierlichen und systematischen Überwachung nach dem Inverkehrbringen können Hersteller gewährleisten, dass ihre Medizinprodukte den versprochenen Nutzen für Patienten, Anwender oder Dritte bringen und das keine unbeherrschten Risiken bestehen.
Quelle:
Abschlussarbeit zum Thema "Erstellung eines Management-Leitfadens zum Umgang mit klinischen Daten im Rahmen des QM-Systems eines Medizinprodukte-Herstellers" von Hicran Karadayi
Was sind die Anforderungen an einen PMS-Prozess gemäß MDR? (Teil 1)